Medical coding https://drive.google.com/drive/folders/1VyetIi1F6e938aLG4SXVktv6-09WaQQh?usp=sharing
BET : https://www.slideshare.net/slideshow/bet-ppt/132105157#52
Deviation : Deviation Management Process in the Pharmaceutical Industry (simplerqms.com)
cGMP : https://simplerqms.com/cgmp/
https://www.linkedin.com/pulse/deviation-pharmaceutical-industry-shouvik-mondal-1f/


ISO classification doesn’t dictate airflow
The amount of air is different in an ISO 6 and ISO 8 cleanroom. This means that the HVAC system must be capable of conditioning more than double the air. However, classification alone isn’t sufficient for calculating the airflow.
ISO 14644-1:2015 does not specify the air changes per hour (ACH) for each cleanroom class because it depends on many factors. Air changes per hour is the number of total replacements of a room’s air in one hour. ISO 14644-1:2015 can only tell you the result that you must aim for: the maximum concentration limits for particles. For example, for ISO 7, particles smaller than 0.5 microns (≥0.1 µm, ≥0.2 µm, ≥0.3 µm) are not taken into consideration. The concentration of particles of ≥0.5 µm should be below 352,000; particles of ≥1 micron should be below 83,200; and particles of ≥5 microns should be below 2,930.
The ISO cleanliness level (ISO 8, ISO 7, ISO 6 and ISO 5) however gives a hint on the ACH range required. Notice that the term “range” is used—not “value”. A cleanroom with activities that generate few particles versus one that generates a lot of airborne particles, even if both ISO 7, will not require the same air changes per hour.
Various recommendations for air change ranges can be found on the Internet. At Mecart, using our cleanroom airflow calculator, we assumed 10 to 30 air changes per hour (ACH) for an ISO 8; 30 to 65 ACH for an ISO 7; 80 to 150 ACH for an ISO 6; 200 to 450 ACH for an ISO 5. If there is a significant generation of particles in the process, the higher number in the range is selected. This is a rule of thumb only. The air changes per hour and CFM must be calculated by an HVAC engineer based on experience and understanding of the particle-generating potential of the process.
Certification
As mentioned above, cleanroom certification is based on ISO14644-1, “Classification of air cleanliness by particle concentration” standards. The specifics of the assessment may vary slightly for FDA or EU GMP regulations, but the underlying methodology is standard.
Certification demonstrates that the entire area meets a specific ISO classification by particle concentration. That is, irrespective of the final use of the room, only the design and implementation of the filtration system are considered. The international standard means that a cleanroom tested to meet compliance for ISO 5 standards will meet that standard independent of geography and regulatory aspects (i.e. FDA or EU GMP). This provides a universal standard to show that a cleanroom level has been established. Particle Measuring Systems’ products, including the Airnet II and IsoAir 310P Aerosol Particle Sensors, comply with new ISO standards set in 2015. The interactive software of the Lasair III Aerosol Particle Counter can even step the user through the certification process.
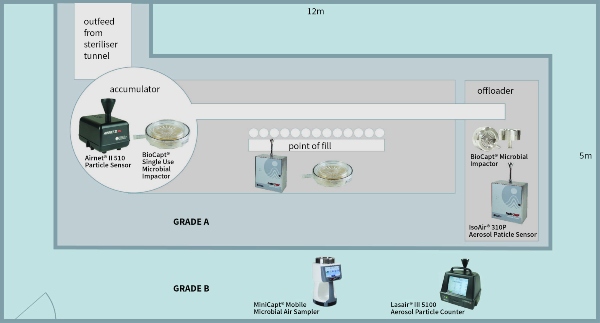
There are many different resources to prove ISO compliance and this article will not cover these in depth. However, using the example of a classic filling machine (Grade A/ISO 5) within a Grade B (ISO 7) background area, the basic rules of testing can be demonstrated.
1. The number of sample points is based on a statistical function of the area:
- Calculate the area of Grade A/ISO5. Find the number of required sample locations in the table.
- Calculate the area of Grade B/ISO7. Find the number of required sample locations in the table.
2. Sample point placement for the Grade A (ISO5) area:
- The sample points must all be equidistant and at work height, irrespective of the activity at the location of their placement.
- Samples are taken in a grid pattern at the identified locations. Derive the minimum number of sampling locations, NL, from ISO 14644-1 Table A.1. This table provides the number of sampling locations related to the area of each cleanroom or clean zone to be classified, and provides at least 95 % confidence that at least 90 % of the cleanroom or clean zone area does not exceed the class limits.
- PASS/FAIL criteria are calculated for ISO and EU GMP Annex 1. It is recommended to have both sets of data, as the FDA requires ISO14644-1, and the EU requires Annex 1 data points (although the EU data would suffice for the FDA).
3. Sample point placement for the Grade B (ISO Class 7) area:
- Repeat the steps used for the Grade A (ISO Class 5) area.
- It may be more difficult to determine the locations of the sample points due to the unusual shape of the room. Derive the minimum number of sampling locations, NL, from ISO 14644-1 Table A.1. This table provides the number of sampling locations related to the area of each cleanroom or clean zone to be classified, and provides at least 95 % confidence that at least 90 % of the cleanroom or clean zone area does not exceed the class limits.
4. A final report is created and marks the end of the certification phase.
Qualification
The qualification phase considers the risks to the quality of the finished product. Each activity must be considered and measured. Continuing with the example of the filling line, let us consider the accumulator table at the exit of the steriliser tunnel. The risk is that glassware (vials/bottles) are exposed to the open environment and operator. Therefore, contamination can fall into clean vials/bottles prior to filling. Operator intervention and the shifting of glassware cause turbulent air movement on the table, impacting contamination risk to the exposed vials/bottles. Therefore, it is an area of contamination risk and the following actions should be taken:
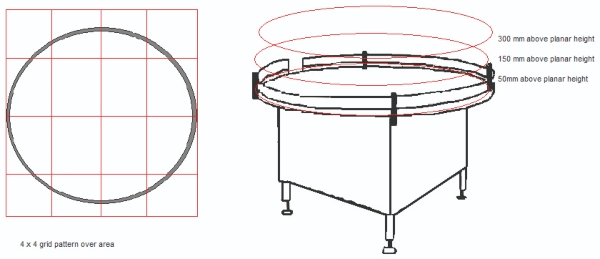
1. Divide the area of risk into a 3x3 or a 4x4 grid. If the activity can occur at several levels, then each level (that is, working height, +150 mm from work height and +300 mm from work height) must be considered.
2. Take a particle sample at the centre of each of the grid squares and on each level.
- Samples are taken during ‘At Rest’ and ‘Operational’ states. It may be required to work around an activity or operator to gain suitable data.
- Slight movement of sample points within the grid square is acceptable. A location is invalid if found to impede normal activities.
3. When all samples are taken this will provide a particle map of the pharmaceutical activity.
Each of the key functions within the cleanroom (filling point, stoppering, general background activities, etc) should be analysed accordingly.
Monitoring
The location of the monitoring points must be based upon a formal risk assessment using tools such as Failure Mode and Effects Analysis (FMEA), Failure Mode, Effects & Criticality Analysis (FMECA), etc, with data from the certification and qualification testing. Other factors, such as equipment interference, mounting points, operator impedance and operator intervention contribute to selecting the final location for the sample probe. In the current regulatory environment, a risk assessment is absolutely required. Without one, poor or incorrect sampling methodology can lead to data unreliably associated with the process or potential impact on finished product quality. Without the option of correlating events, the lack of connection between location and sample frequency can lead to long investigations for out of tolerance events.
There are several steps to defining a risk-based environmental monitoring plan:
1. Process understanding: You must study personnel and material flows within the assessed area in addition to the production operations. This will give an understanding as to how the system is used and what evidence there is to support its state of control, such as:
- Current monitoring practices
- Historical data
- Smoke studies
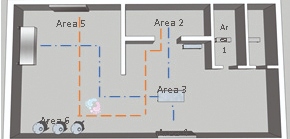
This Gemba walk of the process and rooms is necessary to define the scope of monitoring required and to aid in applying a process that fits with an organization's internal practices. The above figure is an example.2. Definition of critical areas: Identification using Hazard Analysis Critical Control Point (HACCP) helps which critical areas require environmental monitoring, and identifies areas which meet the needs of a critical sample location.
3. Evaluation of sampling methods: You need to make a determination between traditional methods such as volumetric air samplers, newer technologies such as Rapid Microbiological Methods, or manual collection techniques such as swabbing and contact plates. Also, determine if the chosen method needs to be portable, continuous, remote, etc.
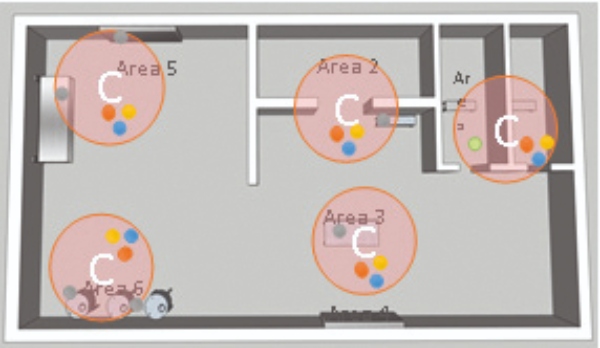
4. Definition of potential sample locations: Determine a single sample location within a critical area, following these criteria (as shown in the adjacent figure):
- Check the available space around the critical area.
- Measure the size of probes and plate holders.
- Determine the accessibility to the location for
operator maintenance. - Assess the interaction between the process operation with personnel and material flows.
- Calculate the probability of potential contamination events.
5. Definition of critical control points (CCP): Each individually considered location is evaluated according to the FEMA method to rank and identify critical sample locations.
6. Define sampling parameters: The sample frequency is found based on the criticality of operations, along with any additional criteria such as incubation parameters, and mitigating measures that might be put into place prior to establishing a monitoring plan.
Sampling practicalities include elements such as the isokinetic sample probe should face into the air stream and the minimum length of tubing should be used. Although different manufacturers claim specific lengths of tubing can be used with their particle counter, this is typically a function of vacuum pump dynamics, and not of particle transportation. Particles of 0.5 μm move freely in long lengths of tubing. However, 5.0 μm particles do not have this same mobility. As 5.0 μm particles are a greater concern, the tubing should be maintained at its shortest recommended lengths1. Particle Measuring Systems quotes maximum tubing lengths based upon the same conditions of airflow, and has a recommended maximum length of 3 m. However, for pharmaceutical particle systems we advise a reduced recommended length of 2 m to ensure transportation of the larger particles.
From the FDA’s Aseptic Processing cGMP Guideline: “Air in the immediate proximity of exposed sterilized containers/closures and filling/closing operations would be of appropriate particle quality when it has a per-cubic-meter particle count of no more than 3520 in a size range of 0.5 μm and larger when counted at representative locations normally not more than 1 foot away from the work site, within the airflow, and during filling/closing operations. This level of air cleanliness is also known as Class 100 (ISO 5).”
The frequency of sampling should reflect the risks and follow from the FDA guidelines on sterile manufacturing and the EU GMP Annex 1. Particle monitoring should be automated and maintained in a continuous state when glassware and products are exposed
operator maintenance.
What Is a Deviation?
A deviation is any departure from approved processes, procedures, instructions, specifications, or established standards.
In the pharmaceutical industry, deviations can occur during drug product development, manufacturing, labeling, packing, sampling, testing, storage, distribution, and other processes.
The deviation definition can vary slightly depending on the regulatory authority and requirements governing the company’s processes.
Some definitions of deviations according to different regulatory organizations and requirements are as follows.
International Organization for Standardization (ISO):
According to ISO 9001:2015, deviations in the context of quality management systems refer to the positive or negative effect resulting from a divergence from the expected or established standards.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH):
In the ICH Q7 Good Manufacturing Practice (GMP) guide for active pharmaceutical ingredients, a deviation is defined as a departure from an approved instruction or an established standard.
Food and Drug Administration (FDA):
In the FDA’s guidance on quality management systems for the pharmaceutical industry and current Good Manufacturing Practice (cGMP) regulations, a deviation is described as a result that falls outside the expected range or fails to fulfill a specific requirement.
What Is Deviation Management?
Deviation management is the systematic process of identifying, assessing, and addressing any deviations that occur from approved instructions or established standards within a company.
The objective of deviation management in the pharmaceutical industry is to promptly identify, investigate, and resolve any deviations.
Deviation management is an essential QMS process in a Pharmaceutical Quality Management System.
By promptly identifying and addressing deviations, pharmaceutical companies can provide safe, uniform, high quality, and effective products to patients while maintaining their reputation in the market and compliance with regulatory requirements.
Effective deviation management plays a crucial role in handling diverse types of deviations, streamlining processes, and fostering continuous improvement.
What Are the Different Classification Types of Deviation?
There are two main classification types of deviations: planned and unplanned deviations.
Planned Deviation
Planned deviations refer to pre-approved and intentional deviations from standard procedures or processes.
These deviations are planned and justified in advance, serving various purposes, such as process improvement, method validation, and temporary process changes.
Some examples of planned deviations include:
- Implementing a temporary manufacturing process change to test and validate potential efficiency improvements.
- Using an alternative raw material as part of a trial to assess its suitability without affecting product quality.
- Temporarily adopting an alternative testing method to validate its accuracy and reliability compared to the standard method.
Change control and change requests are essential components of managing planned deviations in a controlled and systematic manner.
Change request documents are part of the change control process. They provide a formal and structured way to propose and justify the planned deviation, outlining its purpose, scope, and potential impact.
The change control process in the pharmaceutical industry ensures that relevant personnel can assess and approve the proposed change, considering its implications on product quality, safety, and regulatory compliance.
Implementing planned deviations through these processes helps maintain control, traceability, and well-structured documentation.
Unplanned Deviation
Unplanned deviations refer to a departure from approved procedures without prior notice or intention.
Various factors, such as equipment malfunction, employee error, environmental events, or others, can cause them.
Unplanned deviations can significantly impact product quality and safety, and such deviations should be investigated promptly to identify the root cause and prevent them from happening again.
Some examples of unplanned deviations include:
- The sudden breakdown of manufacturing equipment leads to deviations from standard operating conditions and affects product quality.
- Deviations are caused by unforeseen events like power outages, extreme weather conditions, or natural disasters that disrupt manufacturing.
- Accidental introduction of foreign materials into the production process, leading to material contamination and deviation from product specifications.
Companies employ Corrective and Preventive Action (CAPA) processes to effectively address and resolve unplanned deviations.
When an unplanned deviation occurs, the CAPA process is used to implement corrective actions, aiming to mitigate the immediate deviation impact. Afterward, preventive actions are developed to avoid the recurrence of similar deviations in the future.
The CAPA process implementation varies based on each specific company’s established procedures. Companies assess the severity and categorization of the deviation based on risk to determine whether the CAPA process will be initiated.
How Are Deviations Categorized Based on Risk?
Deviations are categorized based on risk levels to assess their potential impact on product quality, patient safety, and the regulatory requirements companies are subject to.
Companies can have different criteria for defining deviation categories, which are well-described in their protocols and procedures. The categorization of deviations may vary based on the nature of the product, industry requirements, and the company’s risk management needs.
The risk-based deviation categories usually include:
- Incidents
- Minor deviations
- Major deviations
- Critical deviations
Incident
An incident is an unplanned or uncontrolled event that does not directly affect the manufacturing process parameters or product quality.
Incidents are generally unrelated to GMP requirements and are often not considered deviations.
However, an incident affecting product specifications can be categorized as a cGMP-related nonconformance.
This categorization varies between companies and must be clearly specified in the company’s procedures.
Examples of incidents are listed below:
- Accidental material spillage occurred in the cleanroom.
- Personnel not appropriately dressed inside the production area.
- A minor typographical error in the documentation that does not affect the overall process or product outcome.
Minor
A minor deviation involves divergences from established procedures or standards that have a noticeable but limited impact on product quality or regulatory compliance.
Examples of minor deviations are listed below:
- A slight variation in the size or appearance of a product that does not affect its efficacy or safety.
- A deviation in the production timeline that leads to a slight delay but does not compromise the overall product quality.
- Documentation errors that do not compromise data integrity.
Major
A major deviation indicates a significant departure from established procedures or standards that can considerably impact pharmaceutical drug quality and regulatory requirements.
Examples of major deviations are presented below:
- A deviation in the manufacturing process results in an out-of-specification (OOS) product requiring investigation and corrective actions to prevent further occurrences.
- A failure of critical equipment during production leads to a significant delay in the manufacturing process and a potential impact on product quality.
Critical
A critical deviation denotes a severe nonconformance from established procedures or standards that poses meaningful risks to product quality, patient safety, or compliance with applicable requirements.
Examples of critical deviations are given below:
- Contamination of a critical raw material used in the production process that may lead to a serious health risk for patients.
- Issues in the packaging process that result in incorrect labeling or dosage instructions which can pose a high risk of incorrect medication intake and patient harm.
- The failure of the HEPA filters in the cleanroom that may lead to potential contamination risks and compromise the integrity of the controlled environment.
When an unexpected event occurs, pharmaceutical companies need to categorize the event to be able to follow the appropriate procedures to solve the issue.
An example of a decision pathway that can be used to categorize incidents and deviations is provided below.
The first step is to identify if the event has a significant impact on the manufacturing process parameters, SOPs, or cGMP. If the event does not have a significant impact, then the event is considered an incident.
However, it is considered a deviation if the event significantly impacts and affects a product attribute.
To determine the severity of the deviation, it is necessary to know if it affects the operations or critical parameters and equipment or instruments.
The deviation can be categorized as minor if none of those are affected. Otherwise, it can be considered a major or critical deviation.
The flowchart below illustrates the deviation categorization process.

What Are the Deviation Management Guidelines in the Pharmaceutical Industry?
Deviation management guidelines vary depending on the specific market that pharmaceutical companies operate in and their product.
The deviation management process must align with the applicable requirements and industry best practices.
In the pharmaceutical industry, various standards, regulations, and guidelines outline requirements regarding deviation management. Below are some of them.
NOTE
Since the list of standards, regulations, and guidelines provided below is not exhaustive, it is essential to consult the applicable requirements for your company for official information.
ISO 9001:2015
The ISO 9001:2015 is a general quality management system standard that specifies requirements for quality systems in several industries. Some pharmaceutical companies choose to comply with ISO 9001:2015 standards.
The standard addresses deviation management in Section 10.2. When a deviation occurs, companies must react to the problem by taking control of the situation and investigating the root cause to correct the problem and manage its consequences.
Preventive actions should be implemented to avoid the recurrence of the deviation, considering similar issues. Companies need to keep records of the deviations, actions taken, and the results of corrective actions.
FDA 21 CFR Part 211
The 21 CFR Part 211 outlines the cGMP requirements for finished pharmaceuticals for companies in FDA-regulated industries.
According to 21 CFR 211.100, pharmaceutical companies must have written procedures for production and process control deviations.
Deviation documents should be drafted, reviewed, and approved by relevant departments and the quality control unit. Procedures must be followed during production, and any deviation from them must be recorded and justified to maintain product integrity and compliance.
ICH Q7
The ICH Q7 defines the GMP guidelines for active pharmaceutical ingredients (API).
Within ICH Q7 quality management guidelines, Section 2.16 emphasizes the importance of documenting and explaining any deviation from established procedures.
The quality documentation related to deviations ensures transparency and accountability in the manufacturing process, helping to maintain the quality, safety, and consistency of API production.
EU and PIC/S GMP Guide Part 1
In the Eudralex GMP and PIC/S GMP Guide Part I for medicinal products, Section 1.8 (vii) specifies that significant deviations from established procedures must be thoroughly recorded.
These deviations should be investigated comprehensively to identify the root cause. Based on the findings, appropriate corrective and preventive actions must be implemented to prevent similar deviations from occurring in the future.
(EU) No 1252/2014
The European Union regulation 1252/2014 sets forth the principles and guidelines of GMP for active substances for medicinal products.
It is established in Article 7 that during the manufacturing process, all quality-related activities, including deviation management processes, must be documented as they are performed. So, if any deviation occurs from the written procedures, it must be documented and explained.
Furthermore, if any deviations impact the quality of the active substance or prevent it from meeting the specifications, a thorough investigation must take place. The findings and conclusions of the investigation must be documented.
There are common requirements across all deviation management regulations, standards, and guidelines that outline the typical steps in the deviation management process.
What Is a Deviation Management Process Flow?
A typical deviation management process flow includes:
- Detection: Identifying the deviation.
- Documentation: Recording the deviation details.
- Investigation: Analyzing the root cause.
- Impact Assessment: Evaluating the impact on product quality.
- CAPA Implementation: Taking corrective and preventive actions.
- Review and Approval: Final review and approval of the deviation management process12.
What Is the Role of Deviation Management Software in Streamlining the Deviation Management Process?
Deviation management software helps streamline the deviation management process by:
- Automating Documentation: Ensuring accurate and consistent documentation.
- Facilitating Investigations: Providing tools for root cause analysis and impact assessment.
- Tracking CAPA: Monitoring the implementation and effectiveness of corrective and preventive actions.
- Ensuring Compliance: Helping maintain compliance with regulatory requirements through automated workflows and audit trails
What is cGMP?
CGMP full form stands for “Current Good Manufacturing Practice.”
CGMP refers to the set of regulations established by the United States Food and Drug Administration (FDA).
The cGMP regulations contain minimum requirements for the methods, facilities, and controls used in the manufacturing, processing, and packing of a product.
CGMP applies to the pharmaceutical, food and beverage, medical devices, and dietary supplement industries. The meaning of cGMP lies in its role in maintaining the safety, efficacy, and quality of products throughout their manufacturing process within these sectors.

What Is the Importance of cGMP?
The importance of cGMP lies in ensuring the consistent quality, safety, and efficacy of products in FDA-regulated industries.
By complying with cGMP regulations, companies can maintain uniform and high-quality manufacturing processes, reduce the risk of product defects or contamination, and meet regulatory and customer requirements.
What Is the Objective of cGMP?
The objective of cGMP is to establish a robust framework that governs every aspect of the manufacturing process, from raw materials to final product distribution, to deliver safe and effective products to consumers.
What Is the Difference Between GMP and cGMP?
The difference between GMP and cGMP is the market they apply to and the level of current practices.
Good Manufacturing Practices (GMP) is a set of requirements applied in various industries, including pharmaceutical manufacturing, to ensure products meet their quality standards.
On the other hand, current Good Manufacturing Practices (cGMP) is a specific set of regulations outlined by the FDA for industries operating within the United States. Accordingly, the “C” in cGMP, which stands for “current,” requires companies to use technologies and systems that are up-to-date to comply with the regulations.
Other differences between GMP and cGMP include quality, cost, and standards, as demonstrated in the illustration below.

Quality
Quality is at the core of both GMP and cGMP requirements, ensuring safe, uniform, and high-quality products.
GMP entails adhering to the required industry standards, while cGMP takes an extra step by keeping up with the latest technologies to ensure updated practices.
Cost
Cost is an inherent consideration in both GMP and cGMP compliance.
However, cGMP tends to be more expensive due to the necessity of additional testing and investments in state-of-the-art technologies to meet the latest standards and requirements.
Current Standard
GMP represents the minimum requirements for product manufacturing, whereas cGMP signifies the advanced and up-to-date regulations that apply to the industry.
Now that we have a clear understanding of what cGMP entails, let’s examine its key requirements to ensure quality and compliance in the manufacturing process.
What Are the Key cGMP Requirements?
The cGMP requirements differ based on the type of product they govern.
Below are the key cGMP requirements applicable to some of the larger Life Science industries.
NOTE
In this section, we will explore some of the cGMP requirements across various industries. However, note that this list is not exhaustive. For official and comprehensive information, always refer to the regulations applicable to your company.
21 CFR Part 111
The 21 CFR Part 111 regulation outlines cGMP requirements specific to the manufacturing, packaging, labeling, and holding operations for dietary supplements.
It covers all aspects of the manufacturing, packaging, labeling, and holding of dietary supplements, including the facilities, equipment, personnel, and procedures used.
The aim is to ensure dietary supplements’ quality, purity, and safety throughout their production and distribution.
21 CFR Part 210
The cGMP requirements for the general manufacturing, processing, packing, and holding of drugs are established in the 21 CFR Part 210 regulation.
It encompasses various aspects of drug production, such as equipment, personnel, and procedures, to ensure consistent product quality and safety.
21 CFR Part 211
The 21 CFR Part 211 specifies cGMP requirements for finished pharmaceutical products.
It covers all stages of pharmaceutical manufacturing, from raw materials to the final packaged products, to guarantee their safety, identity, strength, quality, and purity.
Many Pharma companies operating in the US implement a pharmaceutical quality management system that complies with drug-specific cGMPs, such as 21 CFR Part 211.
21 CFR Part 212
The 21 CFR Part 212 regulation provides cGMP requirements specifically for Positron Emission Tomography (PET) drugs.
It ensures the safe and accurate manufacturing of PET drugs, which are radiopharmaceuticals used in medical imaging.
21 CFR Part 225
cGMP for drugs used in medicated feeds for animal health are outlined in the 21 CFR Part 225 regulation.
It ensures that drugs added to animal food are manufactured, processed, and labeled properly to meet safety and efficacy requirements.
21 CFR Part 606
The 21 CFR Part 606 establishes the cGMP for blood and blood components used in transfusions and medical treatments.
The regulation covers the facilities, equipment, personnel, and procedures used to collect, store, and distribute blood and blood components. It focuses on ensuring the safety and quality of blood products to protect recipients’ health.
21 CFR Part 820
The 21 CFR Part 820, also known as the Quality System Regulation (QSR), sets forth cGMP requirements for medical device manufacturers.
It encompasses various quality management aspects, from design and production to distribution, to ensure safe and effective medical devices. You can learn more about this part of regulation in a dedicated article about 21 CFR Part 820 quality management systems.
For more information on medical device QMS systems, check out our article on Medical Device QMS.
Recognizing the shared key areas of cGMP regulations is essential. However, companies must also grasp the specific requirements pertinent to their operations to develop appropriate processes and controls.
21 CFR Part 11 ?
21 CFR Part 11 is a part of Title 21 of the Code of Federal Regulations (CFR) established by the U.S. Food and Drug Administration (FDA). It sets the criteria under which the FDA considers electronic records and electronic signatures to be trustworthy, reliable, and equivalent to paper records and handwritten signatures12.
Key Aspects of 21 CFR Part 11
- Electronic Records: Defines the requirements for creating, modifying, maintaining, archiving, retrieving, and transmitting electronic records.
- Electronic Signatures: Establishes the criteria for electronic signatures to be considered equivalent to handwritten signatures.
- Validation: Requires that systems used to create, modify, and maintain electronic records are validated to ensure accuracy, reliability, and consistent intended performance.
- Audit Trails: Mandates the use of secure, computer-generated, time-stamped audit trails to independently record the date and time of operator entries and actions that create, modify, or delete electronic records.
- Security Controls: Specifies the need for security measures to ensure that only authorized individuals can access the electronic records and signatures12.
Importance of 21 CFR Part 11
Compliance with 21 CFR Part 11 is crucial for pharmaceutical companies, biotechnology firms, and other regulated industries to ensure the integrity, authenticity, and confidentiality of electronic records and signatures. This regulation helps in maintaining data integrity and supports regulatory compliance during FDA inspections and audits12.
Would you like more detailed information on any specific aspect of 21 CFR Part 11, or do you have any particular concerns or questions about implementing these requirements in your work?
What is CAPA?
CAPA or corrective action and preventive action provide a structure for discovering the main cause of problems and resolving those problems. In addition, it verifies the conditions and solutions for future use and then looking for other possible issues and solutions.
Corrective and Preventive Action is a concept inside the Good Manufacturing Practice (GMP) and ISO 13485. CAPA concentrates on the methodical investigation of discrepancies so that their repetition can be avoided.
To guarantee that corrective and preventive actions are operative, the systematic investigation of the failure incidence is essential in recognizing the corrective and preventing actions undertaken.
For instance, a pharmaceutical or medical device company needs to have a system in place that executes corrective actions and prevents action resultant from the examination of non-conformances, product rejections, audits, deviations, governing inspections, and findings, and trends from process performance and product quality monitoring.
To determine the root cause, an organized approach to the investigation process needs to be utilized. The CAPA methodology needs to result in the product as well as process perfection and improved product and process understanding.
What Is the CAPA Process and Why It Is Important?
The CAPA process consists of numerous steps. At the time of the process, each step needs to be executed successfully and submissive to the CAPA program.
It is necessary to ensure that each action taken is documented carefully because proper documentation helps in offering vital data for continuous quality improvement.
In the case of medical devices, CAPA is part of the different areas of the life cycle processes:
- Production & process controls
- Equipment and facility controls
- Material controls
- Records, documents, and change controls
- Design controls
During the CAPA process, the real scope of the problem should be determined along with causes and detection methods.
Why Implementation of CAPA Is Necessary For QMS?
As per FDA 21 CFR 820, ISO 9001:2015, and ISO 13485:2016, execution of corrective and preventive actions is considered as the path towards the enhancement and success of quality management systems.
Note that corrective actions are mainly the actions that are based upon the identification of the problem. Through staff suggestions, document reviews, management reviews, or internal audits, it is easy to identify non-conformance internally.
On the other hand, customer grievances/suggestions, customer refusals, non-conformities raised in customer/third-party audits & recommendations by the auditors are some of the external sources that assist in finding the root cause of the problem.
Corrective Action VS Preventive Action
Corrective action is a reaction to all the causes or non-conformance that are stated above and is divided into two phases of action:
- Identification of root cause – To recognize the root cause, Total Quality Management (TQM) tools like Fishbone or Cause & Effects analysis can be utilized. Note that CAPA is going to be appropriate and effective only if you have effectively recognized the root cause of the problem.
- Taking important actions – Addressing the root cause requires important and quick actions. By using an efficient approach of the PDCA (plan–do–check–act or plan–do–check–adjust) cycle, the effectiveness of the corrective action requires to be proved periodically.

Preventive action is a prophecy of the problem and a trying method to prevent the occurrence through self-initiated actions and analysis that are related to your products or processes.
Preventive actions can be introduced with the assistance of active participation of staff members/workers through:
• Improvement teams
• Management review
• Enhancement meetings
• Customer reactions
Such an approach not only assures business growth, decreases rejections but also uses the equipment effectively.
Effective CAPA Implementation Tips
To execute the CAPA process effectively from the beginning, you should first address the problems like lack of adequate documentation and lack of adequate actions. The problems should be addressed as early as possible.
Here are some tips that can help you to implement CAPA in a better way:
- By following the guidelines create a CAPA process quickly
- To ensure better control, don’t forget to keep the data centralized
- Make sure to keep your process simple to assure it’s easy to follow
- At the very first stage of a CAPA process, you need to execute risk management
Clearly distinguish the symptoms from causes and have a clear idea about the problem so that you can reach its root cause more easily.
Steps of CAPA for Life sciences Industry
In seven basic steps, execution of a corrective or preventive action that is capable of meeting quality assurance and regulatory documentation requirements is done.
Identification
This is the first step that involves the identification of the problem. It is necessary to define the situation properly and completely as it exists now. This needs to include the source of the information, complete clarification of the problem, and accessible evidence that the problem exists.
A sample form is offered a corrective/preventive action request that can be used to initiate a CAPA action and gather the initial information.
Recommended Reading: Corrective Action and Preventive Action (CAPA) Form
Evaluation
The situation must be evaluated properly to determine the need for action and then the level of action needed. It is necessary to determine the possible impact of the problem and the real risks linked to the company or customers.
A remedial action plan can be used to explain the steps that need to be taken to prevent any kind of additional adverse effects.
Investigation
A written process for doing an investigation into the problem is formed. This kind of written plan ensures that the investigation is complete, and nothing is missed. This procedure mainly includes an objective for the action, investigation strategy, and assignment of accountability and required resources.
A sample investigation form is included that investigates the problem and contains the objectives and instructions that are required for investigating.
Analysis
The investigation process is used for researching the cause of the problem. The main motive of this analysis is to govern the root cause of the problem defined. Along with this, the contributing cause is also recognized.
A problem analysis form is included which helps record the information which is related to the analysis of the issue.
Action Plan
By using the results from the analysis, one of the best methods for correcting the situation and preventing a future occurrence is better determined by the action plan.
You need to know that tasks that are needed to correct a problem and avoid its reappearance is recognized and included in an action plan.
This type of plan includes changes that need to be made and allocates responsibility for the tasks. With this plan, it becomes easy to recognize a person or person who is accountable for completing each task.
Implementation
The correction and preventive action plan created should then be effectively executed. You need to know that all the required tasks that are listed and described in the action plan are introduced, accomplished, and documented.
Follow-Up
In the CAPA process, one of the most essential steps in completing an evaluation of the actions that were taken. This evaluation should validate the successful completion of the recognized tasks and evaluate the suitability and efficiency of the actions taken.
Once the follow-up is done, the CAPA is complete. It needs to be dated and signed by the authorized personnel.
Therefore, CAPA plays a vital role in the quality management system and improves its effectiveness. Apart from this, CAPA methodology also results in the product as well as process enhancements and ensures a better understanding of the product and process.

In pharmaceutical and biopharmaceutical manufacturing, it is often seen that somewhere in the line something could go wrong and have an effect on the quality. When this happens, the quality issue needs to be resolved in an effective and compliant manner.
In case, the issue is small and there is a solution that can be performed directly and appropriately then the quality event can be closed with an effective correction easily. However, if the problem is substantial then it will be escalated into a CAPA.
The CAPA best practices can help in firming clinical research not only for compliance purposes but at the same time aid accelerate time to market.
Recommended Reading
- Corrective and Preventive Action (CAPA) Procedure
- Clinical Quality Management System [Role of an eQMS]
Impact of Risk Management on CAPA
If you are a medical device manufacturer then risk and risk management are synonymous within your daily operations. Medical device manufacturers must bring a device to the market that will not only prove to be functional for a patient but is also safe and effective to use.
In the medical device industry, risk management is a process that is going to stay for a long time. FDA works to ensure that there is stronger risk management in which the manufacturers of the medical device need to have a clear document process for corrective action and preventive action (CAPA).
Proper CAPA processes for corrective action and preventive action (CAPA) is an important step for medical device companies implementing ISO 13485:2016. Therefore, the concept of a risk-based CAPA process has turned out to be foundational for the success of medical device companies.
Recommended Reading: What Is CAPA in the Medical Device Industry?
CAPA Management Software
As you know, for life science organizations, maintaining full compliance with regulatory requirements, those set by ISO and the FDA are critically important to succeeding.
An integrated CAPA software solution can help companies streamline CAPA management processes. Which typically results in enhanced product quality and safety, increased customer satisfaction, and especially, ensure compliance with the regulations, when implemented properly.
With the help of CAPA management software, it is easy to:
- Recognize and initiate a corrective and preventive action process
- Recognize trends
- Conduct or link to a prevailing investigation and root cause analysis
- Describe action plans to modify or improve
- Assure efficacy checks with an organized verification and closure
CAPA software offers risk assessments and notifies you about the possible problems that might take place. This further permits you to changes that are required before an issue takes place.
When the problems occur, the software assists with root cause analysis to know why the issue happened and what steps can be taken to fix the problem effectively.
CAPA software is mainly used by any business that makes use of a quality control system. One of the major uses for CAPA software is by manufacturers who need to ensure that their processed and products successfully meet the regulatory ethics of their industry.
To summarize, here are some benefits of using CAPA management software:
- Recognize product and process problems along with root cause analysis
- Successfully solve product quality & process problems
- Decrease risk and avert quality issues
- Attain valuable insights into quality problems and possible risks
- Sustain regulatory compliance for your business
Various Electronic Quality Management System vendors provide CAPA Management Software System as one of the modules of their overall solution. Although, there is no one size fits all solution that would suit every life science organization.
SimplerQMS offers a ready-to-use eQMS in which you can easily manage corrective action and preventive action (CAPA) processes. Furthermore, it seamlessly integrates with other subsystems and critical processes, including:
- Customer complaints
- Audit Management
- Change Control
- And others

To see our CAPA Management Software in action and learn how SimplerQMS can help your organization achieve better CAPA management processes book a free demo today!
What is Media Fill?
Media fill is a quality control test in pharmaceutical manufacturing, particularly in sterile production (e.g., injectables, vaccines).
It simulates the production process using a nutrient-rich growth medium (e.g. soybean casein digest broth) instead of the actual product.
Purpose:
1. Evaluate sterility and aseptic processing
2. Detect contamination risks
3. Validate process controls
4. Train personnel on aseptic techniques
Process:
1. Prepare growth medium (simulating product)
2. Fill containers (vials, syringes, etc.) with medium
3. Mimic production process (e.g., filling, capping, labeling)
4. Incubate filled containers for 14-28 days
5. Monitor for microbial growth (contamination)
Acceptance criteria:
1. No growth (sterility) in ≥ 95% of containers
2. Low contamination rate (< 0.1%)
Media fill types:
1. Process simulation (full-scale production)
2. Personnel qualification (operator training)
3. Facility qualification (cleanroom validation)
Media fill ensures the sterility and aseptic processing of pharmaceutical products which give an assurance to the patient safety.
Question and Answer Part -1
Question – 1: What is Media fill f Aseptic Process Simulation (APS)?
Answer:
A media fill or Aseptic Process Simulation (APS) is the performance of an aseptic manufacturing procedure using a sterile microbiological growth medium, in place of the drug solution, to test whether the aseptic procedures are adequate to prevent contamination during actual drug production.
To evaluate the capability of aseptic processing activities, using microbiological growth-promoting media in place of product
The media is made to contact all product contact surfaces of the equipment chain, container closure, critical environment, and process manipulations that the product itself will undergo.
Question – 2: What is the frequency of Media fill?
Answer:
Qualification/validation of a new facility or new production process. Media fill is performed after facility, process, equipment, facility decontamination, personnel training, room qualification, and EM program implementation is complete. APS is one of the last steps in the validation process. Typically a minimum of three (3)
are a regulatory expectation.
Semi-annual is a regulatory expectation for a qualified line/ process.
Question – 3: What are the Risk Assessment and Worst-Case Scenarios?
Answer:
The combination of a hazard and its likelihood of occurring and harming the patient. In aseptic processing, this is a loss of sterility assurance.
Risk assessment can be used to determine the worst-case manufacturing scenarios, holistic approach –
- Operating conditions-including personnel
- Interventions
- Container closure – size and configuration
- Line speed
- Batch size
Question – 4: What is the Study Design for Media fill?
Answer:
Ensure that the study/program incorporates risk factors and assesses the state of process control and should simulate manufacturing operations including “worst case” activities and conditions as identified during risk assessment.
Some factors to be simulated/evaluated during Aseptic Compounding
- The entire aseptic compounding process should be simulated including all aseptic additions and can be stand-alone for the compounding step or integrated with filling.
- Filling
- The longest duration of run on an aseptic processing line.
- Interventions – inherent (routine) and corrective (non-routine)
- Aseptic assembly-line set up
- Number of personnel – duration and activities, shift changes, breaks.
- Number of aseptic additions/transfers.
- Connections/disconnections.
- Processes such as Lyophilization.
- Line speed and configuration.
- Container closure types/systems.
- Inert gassing.
- Number of units filled.
- Frequency and number of runs
Question – 5: What is the content of the Media fill protocol?
Answer:
An approved protocol should be in place prior to starting the study.Information/instructions in the protocol should include at a minimum:
- Responsible personnel for execution, and testing.
- Rationale for “worst-case” scenarios.
- Identification of room, filling line, equipment, and process flow.
- Types of container closure to be used
- Fill volume
- Minimum number of units to be filled and rationale.
- Line speed(s)
- Type of media to be used with rationale.
- Number and types of interventions.
- Number, identity, and roles of the personnel.
- Environmental monitoring to be performed.
- Accountability of units filled.
- Incubation conditions and durations.
- Inspection of units – pre-incubation, post-incubation and intermediate.
- Acceptance criteria.
- Conditions of exclusion of vials from incubation (this should be rare).
- Growth promotion.
- Conditions for invalidating/canceling – decision-making authority.
- Personnel training requirements.
- Details about batch records to be used.
- Documentation requirements for the final report
Question – 6: What is the content of the Media fill Batch Records (BMR) as compared to regular BMR?
Answer:
A detailed batch record (BR) written in the same format as the production regular BMR with the same data recording and verification/sign-off requirements should be in place.
An additional section detailing the step-by-step performance of the Interventions should be a part of this BR. All interventions performed (planned and unplanned)with details such as time, operators involved, duration, the identity of the tray filled, any line stoppages, and sample units removed, should be clearly documented.
Question – 7: What should be recorded additional information in the Media fill Batch Records (BMR) as compared to regular BMR?
Answer:
All results should be a part of the batch record including –
- Environmental and personnel Monitoring results
- Number of units filled, number of units incubated, full accountability.
- Number of units rejected pre-incubation with cause for rejection.
- Results of inspection – number of units positive for growth, tray identity, detailed investigation with root cause, and corrective/preventive actions.
- Growth Promotion of media after incubation.
- Record of activities and occurrences during the Amedia fill.
- Any deviations/OOS.
- A Final Report which evaluates the entire media fill and formulates a conclusion on the acceptability is required.
- Quality unit oversight of the entire process including observation in real-time
Question – 8: What is Point to Consider during the Media fill?
Answer:
1. Number of Runs – for a new line/process a minimum of three runs are required. For ongoing requalification – minimum semi-annual. If different processes are performed on the same line, each process has to be re-evaluated semi-annually.
2. Container closure – if multiple sizes of containers of the same type and in the same process are used, a bracketing approach (smallest-largest) may be used.
3. Filling speed – should be set at the production filling range. However, if higher or lower speeds present “worst-case” conditions those may be used.
For example, the Bracketing approach of the highest speed with smallest unit (operational challenge-PM generation) or the slowest speed with largest vial (larger neck size – maximum exposure) may be used.
4. Fill volume- container need not be filled to full capacity. The amount of media should be sufficient to contact all container–closure surfaces when inverted and allow for the detection of microbial growth.
5. Media used – the most common medium used is Soybean Casein Digest Medium (SCDM). This medium is capable of supporting the growth of aerobic microorganisms commonly found in the clean room environment and personnel samples. If the process being validated is anaerobic then Fluid Thioglycollate Medium (FTM) may be used.
6. Inert gassing – Typically Nitrogen or other inert gases are used to protect oxygen-sensitive products and also to provide positive pressure for transfer. Nitrogen should be replaced by Compressed air using the same
method of delivery and at the same steps.
Question – 9: What is the Duration and number of units filled in media fill?
Answer:
Duration and number of units filled – the duration should simulate the longest fill or be representative of routine operations. The duration should be sufficient to allow all interventions and process steps to be executed. Number of units filled during APS should be based on contamination risk and sufficient to simulate the process.
- Generally, 5,000-10,000 units are considered acceptable for average production runs.
- For production batches less than 5,000 units, the APS batch should be equal to the production batch size.
- For production batches 5,000 to 10,000 units, the APS batch should be comparable size (5,000-10,000 units).
- For production batches >10,000 units, the APS batch should be > 10,000 units with several approaches to the batch size and filling process.
Question – 10: What is Interventions in Media fill?
Answer:
Activities performed by personnel in proximity to the aseptic fill zone are called Interventions. Some of these are unavoidable and part of the process.
Interventions in aseptic processes should be kept to a minimum. The Risk Assessment performed should be used to record and evaluate the contamination risk posed to the product due to each intervention.
- Identification of Interventions- The type and frequency of each intervention must be identified. Hence a list of interventions with the frequency of occurrence is to be maintained and re-evaluated.
- The interventions are grouped into two categories – Inherent (routine) and Corrective (non-routine).
Question – 11: What are Inherent (routine) Interventions?
Answer:
Inherent (routine) Interventions-
These are normal planned activities that occur during an aseptic filling process.
Some examples are
- Equipment set up – aseptic assembly
- Fill weight checks and adjustments
- Recharging stoppers and other closures
- Environmental Monitoring sampling
- Shift changes, breaks, duration of personnel activity
Question – 12: What is Corrective (non-routine) interventions ?
Answer:
Corrective (non-routine) interventions
These are performed to correct an aseptic process during execution. They are not a part of the normal aseptic process but they are well defined and recognized as occurring on infrequent occasions.
Some examples-
- Container breakage and picking up fallen units
- Correcting stopper jams
- Changing out filling needles or equipment
- Pulling samples
- Clearing rejected units
- Maintenance work – line stoppage
- Changing out of filters, tubing, and pumps
Question – 14: What is the List and process for Interventions?
Answer:
- The list of interventions is to be re-evaluated at predetermined intervals or if any unusual events occur during production runs.
- There should be established procedures that describe how to perform these interventions.
- Only personnel trained/qualified in the interventions should be permitted to perform them.
- During an APS these interventions should be incorporated to represent the type and frequency of each type on the approved list.
- During routine production operations, any interventions performed should be documented and frequency noted. If any unusual interventions are performed they should be evaluated and a Risk Assessment performed as necessary.
- Based on the risk/evaluation they should be incorporated into the approved list of interventions for APS.
Question – 15: What is Process Qualification?
Answer:
Lyophilized Products – most are aseptically filled and then transferred to a pre-sterilized lyophilization chamber and subject to lyophilization.
The process of loading the partially stoppered units into the lyophilizer, the lyophilization process and the final unloading of the chamber must be captured during an APS.
This can be done as follows-
- Load /unload with shortened hold time and partial vacuum.
- Simulated lyophilization- where the units are held under partial vacuum for the full duration of
the cycle. - In both cases, sterile air is used to vent the chamber instead of Nitrogen.
- APS is used to qualify and requalify the aseptic steps of the lyophilization process.
Question – 16: What is Personnel Qualification?
Answer:
The requirements and the process for the qualification of personnel should be documented in a procedure and results documented and records maintained per each person.
Pre-requisite – all relevant training such as gowning qualification, clean room, aseptic behavior training, GMP training, and procedure training.
Initial Qualification- Personnel should participate in a successful APS in which they perform activities that they would normally perform.
Periodic Qualification- Personnel should participate in a successful APS in which they perform activities that they would normally perform once per year at a minimum.
Disqualification can occur if the personnel fails to participate in periodic qualification, fail gown certification repeatedly, or participate in a failed APS whose failure was directly attributed to their poor aseptic technique.
Question – 17: What is a Pre-incubation inspection?
Answer:
- After the completion of the filling and sealing process, the exact unit count is recorded and verified.
The units are subject to pre-incubation inspection. - The purpose is to remove all non-integral units which would have been removed during normal product
inspection. - Once again count of units proceeding for incubation is documented and verified.
- Some examples of units that may be removed are units with cracks, misaligned or missing stoppers, poor crimps- units with compromised container closures.
- These would also be removed during a routine inspection of the product.
- No unit may be removed due to cosmetic defects.
- The number of units removed and the cause should be documented and verified.
Question – 18: What is the Incubation conditions Media fill?
Answer:
- Incubation conditions should be suitable for the recovery of bioburden and environmental isolates.
- It should be in the range of 20-350 C.
- Incubation time should be not less than 14 days.
- A single incubation temperature for 14 days or two temperatures for 7 days each may be used.
- If two incubation temperatures are used it is recommended to start incubation at the lower temperature.
- Prior to the start of incubation, all units should be inverted or manipulated so that the media comes into contact with all internal surfaces.
Question – 19: What is Post–incubation inspection?
Answer:
- After completion of incubation, all APS units are inspected visually for the presence of microbial growth.
- Personnel trained to detect low/high levels and different types of microbial growth should perform the inspection.
The count of units is performed and verified after inspection. - If non-integral units are found an investigation should be performed as these should have been detected during pre-incubation inspection.
- All these activities should have the oversight of the Quality unit.
- Growth Promotion should be performed after the final inspection.
- In addition to ATCC cultures, the most common environmental isolates should be used.
Question – 20: What are the Acceptance Criteria for Media fill?
Answer:
The target acceptance criteria for the APS study is zero contaminated units.
As Per FDA Guidance for Aseptic Processing
When filling <5000 units – one contaminated unit is cause for revalidation
When filling 5000 to 10,000 units-
- One contaminated unit – investigation and possible repeat media fill.
- Two contaminated units- revalidation following an investigation.
When filling > 10,000 units-
- One contaminated unit – investigation
- Two contaminated units- revalidation following an investigation
Question – 21: What is the Investigation of an APS positive/contamination?
Answer:
A thorough investigation should be performed and documented with the root cause and corrective/preventive actions clearly identified.
At a minimum, the investigation should include the following-
- Species-level identification of the contaminant and comparison to EM/personnel isolates.
- A holistic look at all systems and engineering controls.
- Environmental trend and recovery review.
- Personnel activities and qualifications.
- Sanitization process review.
- Review of batch record, sterilization processes, and any deviations.
- Tracing the location of the contaminant and correlating to any interventions.
- Trend review of previous APS
REFERENCES
1. FDA Guidance for Industry- “Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practices.” September 2004.
2. PDA Technical Report No. 22 – “Process Simulation for Aseptically Filled Products.” 2011.
3. EU GMP Annex 1: Manufacture of Sterile Medicinal Products. 2020
DEFINITION:
“Deviations are the measured differences between the observed and expected or normal values for a product or process condition or a departure from a documented standard or procedure”. A deviation may occur during testing and sampling of finished products and raw materials acceptance and manufacturing. For the sake of continuous improvement and compliance to Good Manufacturing Practice (GMP), if any deviation occurs from the official procedures, then it must be documented. Quality Risk Management (QRM) principals employed in the firm should make sure that all the deviations occurred are rectified and recorded.
Guidelines:
What Is Deviation Management?
- The process of identifying, assessing, and correcting deviations from approved instructions or established standards is known as deviation management. Let us consider the pharmaceutical industry as an example. Your cleanroom's high-efficiency particulate air filters have failed, potentially contaminating one or more batches. This is a critical error that could have serious consequences.
- Many life science organizations have streamlined the entire deviation management process by implementing an Electronic Quality Management System (eQMS) that allows for efficient deviation management.
What Are the Different Types of Deviation ?
In general, your organization will encounter two types of deviations:
- Planned Deviations
- Unplanned Deviations
1.Planned Deviations:
- Planned deviations are temporary deviations from an existing protocol or process that have been pre-approved.
- They are limited to a specific time period or a number of batches. Your organization makes these changes to avoid a potentially hazardous situation.
- Deviations are planned in such a way that they do not compromise the safety and efficacy of your products.
Examples of planned deviations in the pharmaceutical industry:
- Change in batch size due to reduced availability of raw materials
- Change in batch size for a specified number of batches
- Change in supplier for excipients
2.Unplanned Deviations:
- Unplanned deviations are non-compliances with your designed protocols or systems that occur at any stage of a product's lifecycle, including manufacturing, testing, holding, packaging, and storage.
- Unplanned deviations are also referred to as unplanned events, uncontrolled events, or incidents.
- They can be caused by human error, utility failure, equipment/instrumentation breakdowns, or malfunctions.
Examples of unplanned deviations in the pharmaceutical industry:
- Interruption of power supply resulting in equipment breakdown
- Accident at the site due to human error
- Disruption of utility services
What are the Deviation Classification Categories?
There are four deviation classification categories:
- Critical deviation.
- Major deviation.
- Minor deviation.
1. Critical Deviation:
- Deviation that cloud has a significant impact on the production quality or GMP system.
Examples of critical deviation are given below but are not limited to :
- Usage of contaminated raw materials and solvents.
- Failure to process step during the manufacturing.
- Use of obsolete batch document/test method
- Filter integrity failure.
2. Major Deviation:
- Deviations that could have a moderate to a considerable impact on product quality or GMP system.
Ex gave below but not limited to:
- Machine breakdown during processing.
- Mix-ups of cartons of the same product with different strength
3. Minor Deviation:
The deviation will not have any direct impact on the quality of the product or the GMP system.
Examples of minor deviations are given below but are not limited to :
- Minor errors in batch records or documents that do not affect the integrity of the date.
- Spillage of material during dispensing.
- Failure to meet environmental conditions during batch processing
CAPA is a fundamental management tool that should be used in every quality system.
Corrective Actions
- A corrective action is a term that encompasses the process of reacting to product problems, customer complaints or other nonconformities and fixing them. The process includes:
- Reviewing and defining the problem or nonconformity.
- Finding the cause of the problem.
- Developing an action plan to correct the problem and prevent a recurrence.
- Implementing the plan.
- Evaluating the effectiveness of the correction.
Preventive Actions
- A preventive action is a process for detecting potential problems or nonconformance’s and eliminating them. The process includes:
- Identify the potential problem or nonconformance
- Find the cause of the potential problem
- Develop a plan to prevent the occurrence.
- Implement the plan
- Review the actions taken and the effectiveness in preventing the problem.
Differences between Corrective and Preventive Actions:
The process used for corrective actions and preventive actions is very similar and the steps outlined in this document can be used for either. However, it is important to understand the differences and also be aware of the implications involved in performing and documenting each.
1.Corrective Action:
- A corrective action is a reaction to a problem that has already occurred. It assumes that a nonconformance or problem exists and has been reported by either internal or external sources. The actions initiated are intended to: a) fix the problem and b) modify the quality system so that the process that caused it is monitored to prevent a reoccurrence. The documentation for a corrective action provides evidence that the problem was recognized, corrected, and proper controls installed to make sure that it does not happen again.
- To address the Corrective Action clause you should be identifying the root cause of non-conformances that have already taken place and implementing immediate corrective actions to contain the situation and long term corrective actions to prevent their re-occurrence.
2. Preventive Action:
A preventive action is initiated to stop a potential problem from occurring. It assumes that adequate monitoring and controls are in place in the quality system to assure that potential problems are identified and eliminated before they happen. If something in the quality system indicates that a possible problem is or may develop, a preventive action must be implemented to avert and then eliminate the potential situation. The documentation for a preventive action provides evidence that an effective quality system has been implemented that is able to anticipate, identify and eliminate potential problems.
7 Steps of CAPA for Pharmaceutical Industry
Implementing an effective corrective or preventive action capable of satisfying quality assurance and regulatory documentation requirements is accomplished in seven basic steps:
.Identification - Clearly define the problem
.Evaluation - Appraise the magnitude and potential impact
.Investigation - Make a plan to research the problem
.Analysis - Perform a thorough assessment with documentation
.Action Plan - Create a list of required tasks
.Implementation - Execute the action plan
.Follow Up - Verify and assess the effectiveness
1 - Identification - Clearly define the problem
The initial step in the process is to clearly define the problem. It is important to accurately and completely describe the situation as it exists now. This should include the source of the information, a detailed explanation of the problem, the available evidence that a problem exists.
This should include:
- The source of the information
The specific source of the information is documented. There are many possible sources: Service requests, Internal Quality Audit, Customer complaints, Internal quality audits, Staff observations, Trend data, QA inspections, Process monitoring, Risk analysis, Process performance monitoring, Management review, and Failure mode analysis. This information is important for the investigation and action plan, but also useful for effectiveness evaluation and communicating the resolution of the problem.
- Detailed explanation of the problem
A description of the problem is written that is concise - but complete. The description must contain enough information so that the specific problem can be easily understood.
- Documentation of the available evidence that a problem exists.
List the specific information, documents, or data available that demonstrates that the problem does exist. This information will be very important during the investigation into the problem. For example, the evidence for a product defect may be a high percentage of service requests or product returns. The evidence for a potential equipment problem may be steadily increasing downtime.
- Corrective/Preventive Action Request form :
A sample form is provided “Corrective/Preventive Action Request that can be used to
- initiate a CAPA action and collect the initial information.
2 - Evaluation - Appraise the magnitude and impact
The situation must be evaluated to determine both the need for action and then, the level of action required. The potential impact of the problem and the actual risks to the company and/or customers must be determined. Essentially, the reasons that this problem is a concern must be documented.
An evaluation should include:
- Potential Impact of the problem
Determine and document specifically why the problem is a concern and what the impact to the company and/or customers may be. Concerns may include costs, function, product quality, safety, reliability, and/or customer satisfaction.
- Assessment of Risk
Using the result of the impact evaluation, the seriousness of the problem is assessed. The level of risk that is associated with the problem may affect the actions that are taken. For example, a problem that presents a serious risk to the function or safety of a product may be assigned a high priority and require immediate remedial action. On the other hand, an observation that a particular machine is experiencing an increasing level of downtime each month may have a lower priority.
- Remedial Action that may be required
The potential impact and risk assessment may indicate a need for some immediate action to remedy the situation until a permanent solution can be implemented. In some cases the remedial action may be adequate. If so, the CAPA can then be closed, after documenting the rationale for this decision and completing appropriate follow up.
- Remedial Action form
A sample “Remedial Action” form is included. This form should be used to explain the steps that must be taken to avoid any further adverse effects.
3 - Investigation - Make a plan to research the problem
A written procedure for doing an investigation into the problem is created. A written plan helps assure that the investigation is complete and nothing is missed.
This procedure should include:
- The objectives for the action
The objective is a statement of the desired outcome(s) of the corrective or preventive action.
The action will be complete when all aspects of the objective have been met and verified.
- An investigation strategy
A set of specific instructions for determining the contributing and root causes of the problem is written.
This procedure directs a comprehensive review of all circumstances related to the problem and must consider: equipment, materials, personnel, procedures, design, training, software, external factors.
- Assignment of responsibility and required resources
An important part of the investigation procedure is to assign responsibility for conducting each aspect of the investigation. Any additional resources that may be required is also identified and documented. For example, specific testing equipment or external analysis may be required.
- Investigation Procedure form
A sample “Investigation Procedure” form is included. This is a written plan of action for the investigation into the problem. It should include the overall objective and the instructions for conducting the investigation. The person or persons responsible for the investigation and an expected completion date should also be entered.
4 - Analysis - Perform a thorough assessment
The investigation procedure is used to conduct the investigation into the cause of the problem. The goal of this analysis is primarily to determine the root cause of the problem described, but any contributing causes are also identified.
- Every possible cause is identified and appropriate data collected.
- A list of all possible causes is created which then form the basis for collecting relevant information, test data, etc.
- The necessary data and other information is collected that will be used to determine the primary cause of the problem.
- The results of the data collection are documented and organized.
- Data may come from a variety of sources: testing results and/or a review of records, processes, service information, design controls, operations, and any other information that may lead to a determination of the fundamental cause of the problem.The data collected is organized into a useable form.The resulting documentation should address all of the possible causes previously determined. This information is used to determine the root cause of the problem. The effectiveness of the analysis will depend on the quality and thoroughness of the information available.
- Everything related to the problem must be identified, but the primary goal must be to find the root cause.Use the data to complete a Root Cause Analysis. This involves finding the actual cause of the problem rather than simply dealing with the symptoms. Finding the primary cause is essential for determining appropriate corrective and/or preventive actions.
- Problem Analysis form
A sample “Problem Analysis” form is included. This form is optional but is intended to be used for recording information related to the analysis of the problem. The form can be used as a collection point for the information discovered during the analysis and any supporting data or documentation can be attached.
5 - Action Plan - Create a list of required tasks
Using the results from the analysis, the best method(s) for correcting the situation (or preventing a future occurrence) is determined and act ion plan developed. All of the tasks required to correct the problem and prevent a recurrence are identified and incorporated into an action plan.
The plan includes changes that must be made and assigns responsibility for the tasks. The action plan should also identify the person or persons responsible for completing each task.
- Actions to be completed
List all activities and tasks that must be accomplished to correct the existing problem or eliminate a potential problem, and prevent a recurrence. It is very important identify all actions necessary to address everything that contributed to or resulted from the situation.
- Document or Specification Changes
Needed changes to documents, processes, procedures, or other system modifications should be described. Enough detail must be included so it is clearly understood what must be done and what the outcome of the changes should be.
- Process, Procedure, or System changes
If any changes to processes, procedures, or systems must be made they are described. Enough detail should be included so that it is clearly understood what must be done. The expected outcome of these changes should also be explained.
- Employee Training
Employee training is an essential part of any change that is made and should be made part of the action plan. To be effective, all modifications and changes made must be communicated to all persons, departments, suppliers, etc. that were or will be affected.
- Action Plan form
A sample “Action Plan” form is included. This should provide a set of written procedures that detail all of the actions that must be done to resolve the problem and prevent it from recurring. This includes corrective and preventive activities, document changes, training, etc. The person or persons responsible and an expected completion date should also be entered on the form.
6 - Implementation - Execute the action plan
The corrective / preventive action plan that has been created is now implemented. All of the required tasks listed and described in the action plan are initiated, completed, and documented.
- Implementation Summary
All of the activities that have been completed as required in the “Action Plan” should be listed and summarized. This section should contain a complete record of the actions that were taken to correct the problem and assure that it will not recur. This includes changes, preventive measures, process controls, training, etc.
- Documentation
All documents or other specifications that have been modified are listed. Typically the documentation would be attached to a final printed report of this CAPA action. This will facilitate verification of the changes for the follow up.
7 - Follow Up - Verify and assess the effectiveness
One of the most fundamental steps in the CAPA process is completing an evaluation of the actions that were taken.
This evaluation must not only verify the successful completion of the identified tasks, but also assess the appropriateness and effectiveness of the actions taken.
- Key questions
- Have all of the objectives been met? (Did the actions correct or prevent the problem with assurances that the same situation will not happen again?)
- Have all recommended changes been completed and verified?
- Has training and appropriate communications been implemented to assure that all relevant employees understand the situation and the changes that have been made?
- Has an investigation demonstrated that that the actions taken have not had any additional adverse effect on the product or service?
- Verification results
Make sure that appropriate information has been recorded that provides proof that all actions have been completed successfully.
- Validation results
- A validation of the action is done. This must document that:
- The root cause of the problem has been solved,
- Any resulting secondary situations have been corrected,
- Proper controls have been established to prevent a future occurrence,
- The actions taken had no other adverse effects.
- Adequate monitoring of the situation is in place.
- Completion
When the Follow Up has been finished, the CAPA is complete. It should be dated, and signed by appropriate, authorized personnel
Audits And Its Types
Types of Audits in the Pharmaceutical Industry
- Internal Audits: Purpose: It is conducted by the organization itself to assess the effectiveness of its own quality management system and compliance with internal procedures and policies. Frequency: Regularly scheduled (e.g., quarterly, annually) or as needed. Scope: Can cover all aspects of operations, including manufacturing, quality control, and documentation.
- External Audits: Regulatory Audits: Purpose: Conducted by regulatory authorities (e.g., FDA, EMA) to ensure compliance with regulatory requirements such as Good Manufacturing Practices (GMP). Frequency: Typically scheduled or triggered by significant changes or issues. Customer Audits: Purpose: Conducted by clients or customers to ensure that their suppliers or contract manufacturers comply with their quality standards and regulatory requirements. Frequency: Scheduled based on contractual agreements or as needed. Third-Party Audits: Purpose: Conducted by independent organizations or certification bodies to assess compliance with specific standards (e.g., ISO 9001, ISO 13485, ISO 14001). Frequency: Typically, part of certification or recertification processes.
- For-Cause Audits: Purpose: Specific problems, complaints, or incidents that require immediate investigation. Frequency: As needed, based on the occurrence of issues or deviations. Scope: Focused on particular areas of concern or specific processes.
- Supplier Audits: Purpose: Conducted to evaluate and ensure the quality and reliability of suppliers and their adherence to contractual and regulatory requirements. Frequency: Typically scheduled periodically or triggered by performance issues. Scope: Can cover manufacturing processes, quality control, and supply chain management.
- System Audits: Purpose: Focused on the entire quality management system to ensure it is effectively implemented and maintained. Frequency: Regularly scheduled as part of continuous improvement efforts. Scope: Comprehensive review of policies, procedures, processes, and records.
- Product Audits: Purpose: Focused on specific products to ensure they meet quality and regulatory requirements. Frequency: As needed, based on product lifecycle stages or issues. Scope: Examination of product specifications, testing, and documentation.
- Process Audits: Purpose: Focused on specific processes within the organization to ensure they are controlled and effective. Frequency: Regularly scheduled or as needed. Scope: Detailed review of process steps, controls, and records.
Risk Assessment Study for Environmental Monitoring (EM) Programme
What is Risk Assessment in EM?
Risk assessment for an Environmental Monitoring (EM) program is a systematic process used to determine:
- Where to monitor
- What to monitor
- How frequently to monitor
- Which monitoring method to use
The goal is to ensure that locations with the highest contamination risk receive the most monitoring attention.
Why Perform Risk Assessment?
Environmental monitoring locations should not be selected randomly.
A risk assessment helps identify areas where contamination could affect product quality and sterility assurance.
Factors Considered
- Proximity to exposed product
- Personnel activity
- Airflow pattern (Smoke Study)
- Equipment design
- Historical EM data
- Cleaning and disinfection effectiveness
- Utility connections
- Intervention frequency
Risk Assessment Process
Step 1: Identify Critical Areas
Example
Sterile vial filling line:
Areas include:
- Filling needle
- Stoppering station
- Conveyor belt
- Operator working area
- Material transfer hatch
All locations are assessed for contamination risk.
Step 2: Evaluate Risk Factors
Typical Risk Parameters
| Risk Factor | Question |
|---|---|
| Product Exposure | Is product exposed? |
| Personnel Activity | Is operator intervention frequent? |
| Airflow Impact | Is airflow disturbed? |
| Historical EM Data | Any previous excursions? |
| Equipment Design | Difficult-to-clean surfaces? |
Step 3: Assign Scores
Example Scoring
| Parameter | Score |
|---|---|
| Low Risk | 1 |
| Medium Risk | 3 |
| High Risk | 5 |
Example Risk Assessment
Filling Needle Area
Product Exposure
Open sterile product present.
Score = 5
Personnel Activity
Frequent interventions.
Score = 5
Airflow Disturbance
Possible during interventions.
Score = 5
Historical Data
Occasional EM recoveries.
Score = 3
Equipment Design
Complex assembly.
Score = 3
Total Risk Score
5 + 5 + 5 + 3 + 3 = 21
Risk Level = High
Monitoring Decision
- Active Air Sampling
- Settle Plates
- Contact Plates
- Continuous Particle Monitoring
Example 2: Room Corner
Product Exposure
No product exposure.
Score = 1
Personnel Activity
Very low.
Score = 1
Airflow Disturbance
Minimal.
Score = 1
Historical Data
No recoveries.
Score = 1
Equipment Complexity
None.
Score = 1
Total Score
1 + 1 + 1 + 1 + 1 = 5
Risk Level = Low
Monitoring frequency can be lower.
Smoke Study and EM Risk Assessment
Smoke study is one of the most important inputs.
Example
Smoke study shows turbulence near stopper bowl.
Risk:
Particles may enter critical zone.
Action:
- Additional EM location added.
- Increased monitoring frequency.
Interview Point
"EM locations are selected based on smoke study results, risk assessment, personnel interventions, product exposure, and historical trend data."
Historical Data Review
Previous EM trends are reviewed.
Example
Location SP-05:
| Month | Result |
|---|---|
| Jan | 0 CFU |
| Feb | 1 CFU |
| Mar | 2 CFU |
| Apr | 5 CFU |
| May | 7 CFU |
Trend indicates increasing contamination risk.
Action
- Increase monitoring frequency.
- Investigate cleaning effectiveness.
- Review operator practices.
Risk Ranking Example
| Risk Score | Category |
|---|---|
| 1–8 | Low |
| 9–15 | Medium |
| >15 | High |
Practical Example (Grade A Filling Line)
Observation
Repeated recovery of Micrococcus luteus from operator monitoring.
Risk Assessment Findings
- Frequent interventions.
- Inadequate glove sanitization.
- High personnel movement.
CAPA
- Retrain operators.
- Increase glove disinfection frequency.
- Additional personnel monitoring.
Outcome
EM trend returned to normal levels.
Risk Assessment (ICH Q9)
What is Risk Assessment?
A systematic process for identifying, evaluating, and controlling risks to product quality and patient safety.
Example: Environmental Monitoring Excursion
Suppose:
Grade B room limit = 10 CFU
Observed result = 18 CFU
Step 1: Risk Identification
Question:
What can go wrong?
Answer:
Microbial contamination of product.
Step 2: Risk Analysis
Evaluate:
Severity
What happens if contamination reaches product?
Impact:
- Sterility failure
- Patient infection
Severity = High
Occurrence
How often does it happen?
Review:
- EM trends
- Historical data
If recurring:
Occurrence = High
Detectability
Can we detect the contamination?
If EM detected it:
Detectability = Good
Step 3: Risk Evaluation
Combine:
- Severity
- Occurrence
- Detectability
Risk may be Medium or High.
Step 4: Risk Control
Actions:
- Deep cleaning
- Disinfection
- Retraining operators
- Additional EM monitoring
Step 5: Risk Review
Verify:
- EM results returned to normal
- No recurrence
Comments
Post a Comment